
Un equipo internacional de científicos ha logrado un descubrimiento que podría poner al alcance de la mano nuevos tratamientos contra el síndrome de Cushing. Su hazaña fue posible gracias a que reprogramaron la identidad de varias células de la glándula pituitaria e identificaron mecanismos esenciales de programación celular epigenética. El equipo considera que los resultados obtenidos podrían conducir a la identificación de nuevas dianas farmacológicas para el tratamiento contra el síndrome de Cushing. Sus resultados se han publicado en la revista Genes & Development y han sido resumidos por la agencia CORDIS.
Se calcula que el síndrome de Cushing afecta a cerca de 0,9 de cada 10 000 ciudadanos de la Unión Europea. Esta enfermedad se caracteriza por una concentración elevada de la hormona cortisol en la sangre. La generación de esta hormona en exceso se debe a la presencia de tumores pequeños en la glándula pituitaria. En los pacientes afectados, esta anomalía en la producción hormonal puede provocar hipertensión, obesidad, diabetes y osteoporosis.
Los afectados pueden engordar en la zona de la cara y el torso pero no en las extremidades, acumular grasa en la clavícula y la nuca, sufrir hematomas con facilidad, presentar hirsutismo en el rostro y padecer debilidad muscular y ósea y depresión. El síndrome de Cushing es una enfermedad grave de larga duración y en ocasiones mortal debido a complicaciones como las mencionadas y otras de índole psicológica.
«En cerca de un 10 % de los afectados por el síndrome de Cushing descubrimos que los tumores causantes contienen células que expresan la proteína Pax7», explica el doctor Jacques Drouin, director de la Unidad Científica de Genética Molecular del Instituto de Investigaciones Clínicas de Montreal (IRCM, Canadá) y añade que «aún no existe un tratamiento farmacológico efectivo contra esta enfermedad. Este hallazgo podría conducir al desarrollo de un tratamiento hormonal de inhibición del crecimiento tumoral similar al que se emplea en otros tumores pituitarios como los prolactinomas.»
La glándula pituitaria
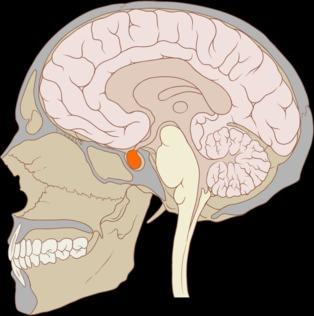
El equipo estudió, bajo la dirección del doctor Drouin, la glándula pituitaria, también llamada glándula endocrina maestra. Ésta se ubica en la base del cráneo, desde donde secreta hormonas que controlan el resto de glándulas del sistema endocrino. Cualquier anomalía de las funciones de esta glándula comporta consecuencias nefastas sobre el crecimiento, la reproducción y el metabolismo.
En la glándula pituitaria cada hormona se genera en células de distinto linaje. Cada una de las identidades celulares se crean mediante programas genéticos de células específicas seguidos durante la fase de desarrollo. El proceso de programación celular es determinante, por lo que es necesario conocer su adecuado funcionamiento para aprovechar los beneficios terapéuticos de la investigación con células madre.
En el trabajo referido se indica que el factor de transcripción Pax7 posee «capacidades pioneras», pues es capaz de abrir la estructura densa de la cromatina en regiones concretas del genoma. Este desenmascaramiento de un subgrupo de las secuencias regulatorias del genoma modifica la respuesta de éste a las señales de diferenciación, de manera que se logra generar distintos tipos de células.
«Reprogramamos la identidad de algunas células de la pituitaria mediante el gen Pax7 con el fin de crear dos tipos distintos de célula», explicó Lionel Budry, antiguo alumno en el laboratorio del doctor Drouin y autor principal del artículo. «Esta maniobra nos permitió mostrar que la proteína Tpit produce un linaje celular distinto en función de la presencia o ausencia de Pax7 y su influencia en la organización de la cromatina.»
En el equipo participaron científicos del IRCM en colaboración con colegas de la Universidad del Mediterráneo (Francia) y el Hospital La Timone de Marsella (Francia) y la Universidad de Utrecht (Países Bajos).
Referencia bibliográfica:
Budry, L. et al., The selector gene Pax7 dictates alternate pituitary cell fates through its pioneer action on chromatin remodeling, Genes & Development, 26 (20): 2299, 2012. doi:10.1101/gad.200436.112.















Hacer un comentario